Bioresorbable Polymer Extrusions of Yarns, Fibers, & Films for Implantable Medical Devices
Bioresorbable Yarns, Fibers, & Films Overview:
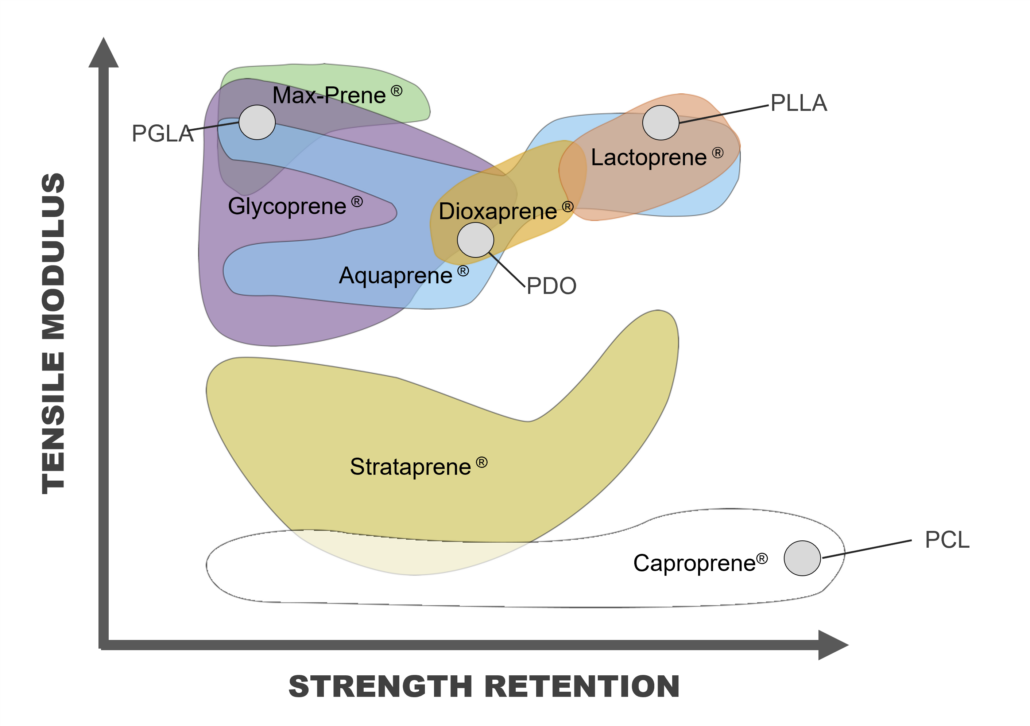
Bioresorbable polymers (also referred to as bioabsorbable polymers, resorbable polymers, absorbable polymers, and degradable polymers) may be processed via melt extrusion into a variety of formats that include but are not limited to monofilament fibers, multi-filament yarns, and films. These extruded materials may be processed further into a final bioresorbable medical device or component that may be implanted in a patient for treatment and safely metabolized or excreted later when the implant is no longer required. Melt extrusion requires a careful balance four (4) main process settings of 1) moisture management, 2) optimizing the polymer melt profile, 3) the appropriate mechanical shear imparted on the molten polymer, and 4) extrudate orientation in-line or off-line. Minimization of moisture ensures polymer degradation is lessened as water is a driving factor in the hydrolytic degradation of resorbable polyesters. Temperature deviations outside of the validated range of an extrusion process may result in generation of increased levels of residual monomer (elevated levels of residual monomer above certain thresholds are associated with cytotoxicity) and degradation of the polymer leading to a decrease average molecular weight (which has the potential to alter a materials degradation profile). A careful balance between mechanical shear and temperature ensures that residual monomer generation is abated while molecular weight of the polymer is maintained. Finally, in-line (or off-line) orientation allows proper sizing and strength setting for extrudate materials. By having well-controlled processes that balance these four (4) factors, extrudate materials can have optimal molecular weight, reduced monomer content, all while ensuring critical material properties of size, strength, and resorption profile are achieved. The section below outlines extrusion processing methods for bioresorbable materials currently available at Poly-Med.

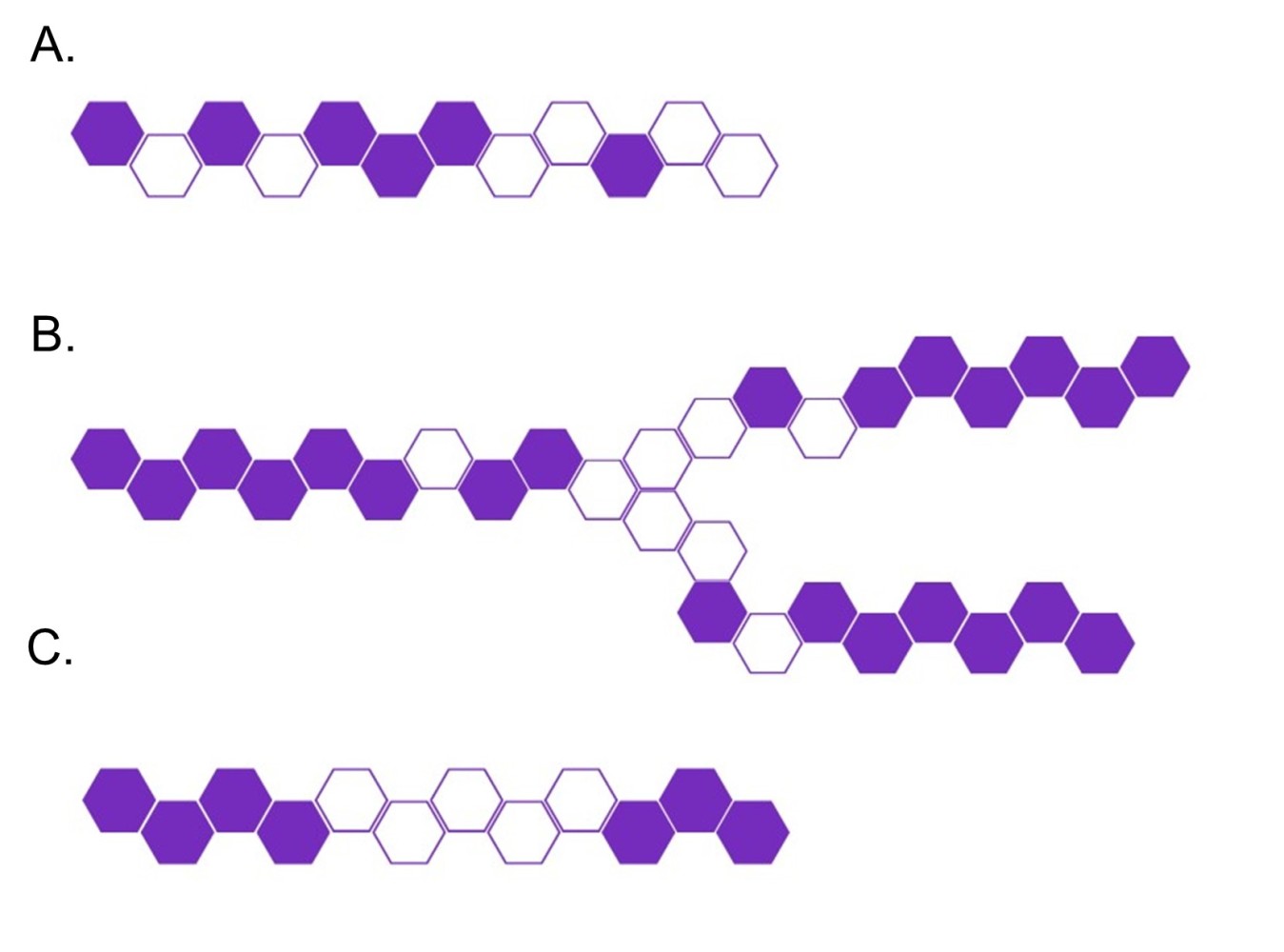
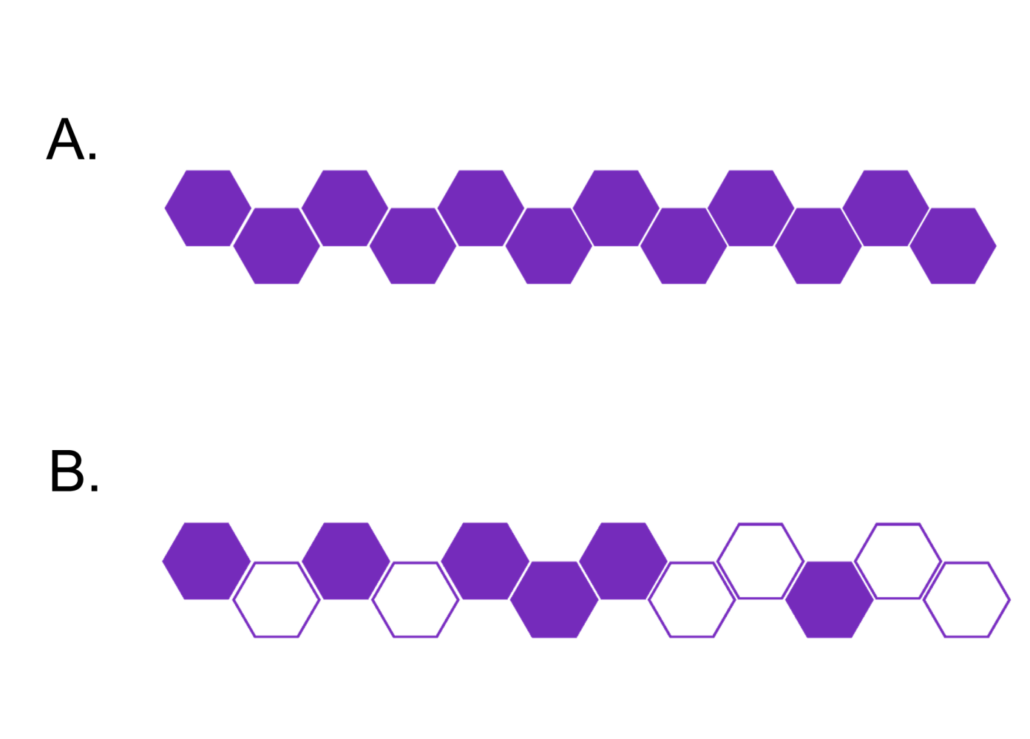
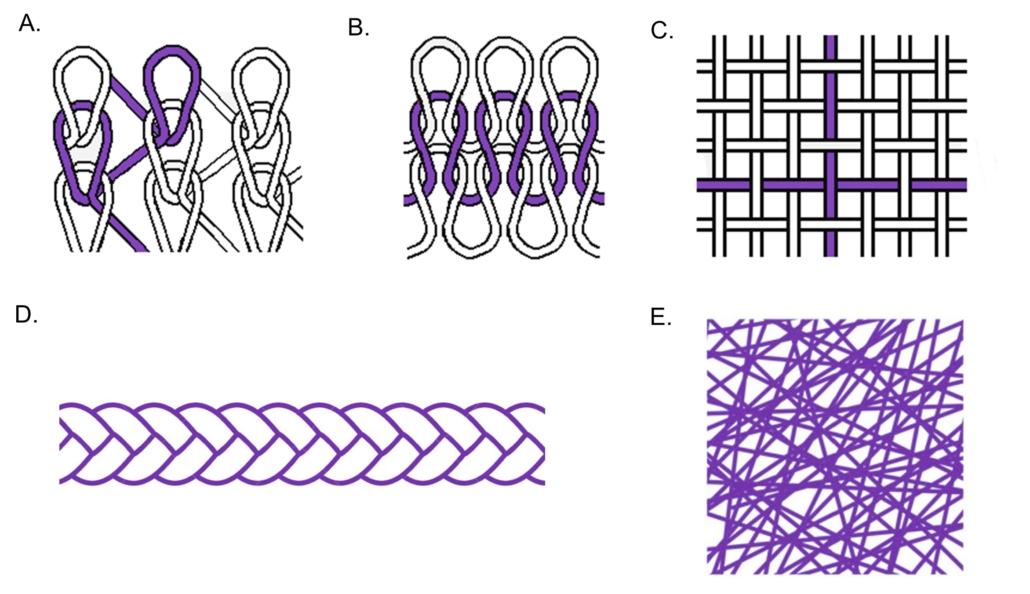
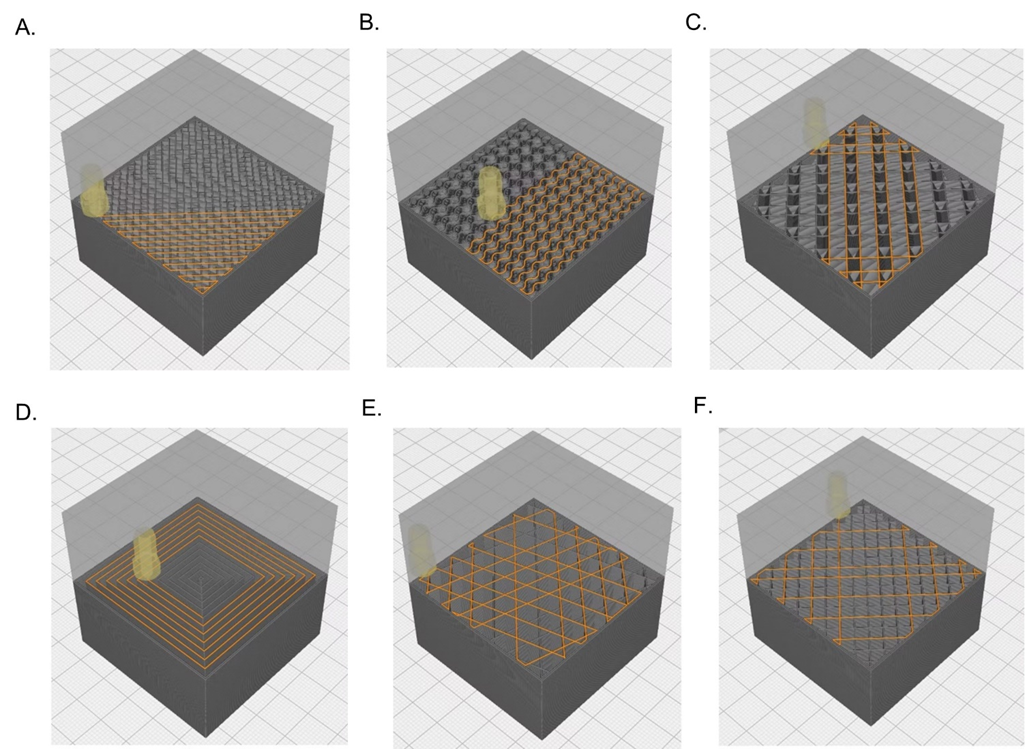
Figure 1: Poly-Med’s advanced absorbable materials may be extruded into monofilament fibers (Panel A), multifilament yarns (Panel B), microfilament fibers (Panel C), and films (Panel D).
Bioresorbable Monofilament Fibers:
Bioresorbable materials may be melt extruded into monofilaments consisting of a single strand of fiber where the diameter is greater than one-hundred (100) microns, or microfilaments consisting of a single strand of fiber with a diameter of less than one-hundred (100) microns. Typically, monofilament fibers are oriented to enhance mechanical properties, dimensional stability, or flexibility. Monofilaments may be utilized as is in smaller diameters as suture materials or for FDM 3D printing in larger diameters. Bioresorbable monofilament fibers may also be processed into biomedical textiles for use in mesh & graft implants or braided into hollow tube structures. Poly-Med offers both custom extrusion of our portfolio of bioresorbable polymers and off-the-shelf monofilament fibers that may be used in downstream processing:
| Material | USP Class | Diameter (mm) | Peak Load (N) | Elongation |
| Max-Prene® 955 | 6-0 | 0.075 – 0.090 | >4 | 15 – 30% |
| Glycoprene® 935 | 5-0 and 6-0 | 0.095 – 0.115 | >5 | 20 – 40% |
| Glycoprene® 7424 + BaSO4 | 3-0 and 4-0 | 0.250 – 0.310 | ≥13 | 20 – 50% |
Bioresorbable Multifilament Yarns:
Bioresorbable materials can be melt extruded into yarns made of multiple strands of filaments ranging from 1.5 – 30 denier per filament and 5 – 80 filaments per bundle. Multifilament fibers are oriented in-line and are known to be utilized as components in biomedical textiles for use in mesh & graft fabrics or braided into suture materials for tissue closure. Poly-Med offers both custom extrusion of our portfolio of bioresorbable polymers and off-the-shelf multifilament yarns that may be used in downstream processing:
| Material | Filament Count | Denier (g/9000m) | Tenacity (gf/denier) | Elongation |
| Glycoprene® 935 | 10 | 180 – 200 | ≥3.0 | >30% |
| Glycoprene® 935 | 20 | 70 – 100 | >2.5 | 40 – 80% |
| Lactoprene® 8812 | 30 | 115 – 125 | >3.3 | 20 – 35% |
| Lactoprene® 8812 | 86 | 160 – 180 | ≥3.0 | 20 – 40% |
Bioresorbable Films:
Bioresorbable materials may also be melt extruded into film formats exhibiting a range of thicknesses and width measurements based on the device dimensional requirements. Poly-Med has the ability to extrude bioresorbable films with thicknesses ranging from seventy (70) to three hundred (300) microns and widths up to 12.7 centimeters. Bioresorbable films may be used as components requiring a degradable layer or as final devices for direct contact with non-structural tissue indications. Poly-Med offers custom extrusion of our portfolio of bioresorbable polymers for development and manufacturing of bioresorbable films.
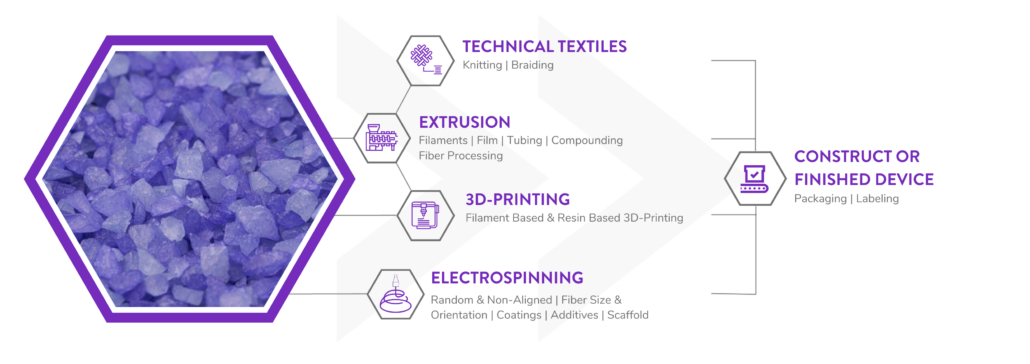
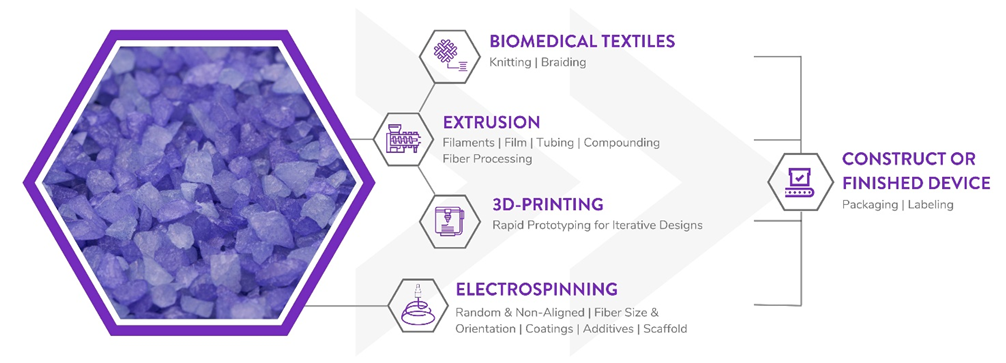
Poly-Med is a Vertically Integrated Bioresorbable Medical Device Manufacturing Partner:
Manufacturing bioabsorbable medical devices is hard. Controlling moisture levels and material degradation through the production cycle requires specialized equipment and process controls to ensure quality of finished devices. In contrast to other bioresorbable polymer manufacturers, Poly-Med produces polymer, is able to extrude this material to desired monofilament or multifilament formats, and is able process this material via warp knitting, weft knitting, or braiding processes to produce custom biomedical textiles. Beyond traditional textiles, we can process our bioresorbable polymers into non-woven formats via electrospinning via our state-of-the-art electrospinning facility. In addition to accessing Poly-Med’s more than thirty (30) years of experience manufacturing bioresorbable polymers, partnering with Poly-Med simplifies your bioresorbable medical device manufacturing supply chain. Contact us to today begin developing your custom bioresorbable medical device product line with a trusted partner for manufacturing advanced bioresorbable medical devices or purchase off-the-shelf high-performance bioresorbable polymers!